Primary vs Secondary Lysosomes: Essential Differences Explained
Have you ever wondered how our cells manage to break down unwanted materials and recycle their components? The answer lies in fascinating organelles called lysosomes. These tiny cellular structures work like the digestive system of our cells, breaking down everything from foreign particles to worn-out cellular components. But did you know there are actually different types of lysosomes? The primary and secondary lysosomes represent different stages in the lysosomal life cycle, each with unique characteristics and functions that are essential for cellular health.
I've spent years fascinated by cellular biology, and lysosomes remain some of the most intriguing organelles. They're like little cleanup crews constantly working behind the scenes to keep our cells functioning properly. Understanding the difference between primary and secondary lysosomes provides crucial insights into how cells maintain themselves and respond to their environment. In this comprehensive guide, we'll explore these differences, their significance, and why these tiny organelles deserve our attention.
What Are Lysosomes?
Before diving into the specific types of lysosomes, let's establish a clear understanding of what lysosomes actually are. Lysosomes are membrane-bound organelles found primarily in animal cells and some plant cells. They function as the digestive system of the cell, containing powerful enzymes capable of breaking down virtually all types of biological polymers—proteins, nucleic acids, carbohydrates, and lipids.
I remember my first time seeing lysosomes under an electron microscope during my biology studies—they appeared as small, dense structures scattered throughout the cytoplasm. What makes them special is their acidic interior, with a pH of around 4.5-5.0, which activates the digestive enzymes they contain. This acidic environment is maintained by proton pumps in the lysosomal membrane that continuously transport hydrogen ions into the lysosome.
The term "lysosome" comes from the Greek words "lysis" (meaning dissolution or destruction) and "soma" (meaning body), quite literally describing their function as "digestive bodies." They were first discovered in 1955 by Belgian cytologist Christian de Duve, who later received the Nobel Prize for this and other discoveries related to the structural and functional organization of cells.
In the cellular life cycle, lysosomes play critical roles beyond just digestion. They participate in various cellular processes including secretion, plasma membrane repair, cell signaling, and energy metabolism. They're also involved in programmed cell death (apoptosis) and can even help defend against pathogens. Understanding the different types of lysosomes helps us appreciate how these organelles accomplish such diverse functions.
Primary Lysosomes: Structure and Function
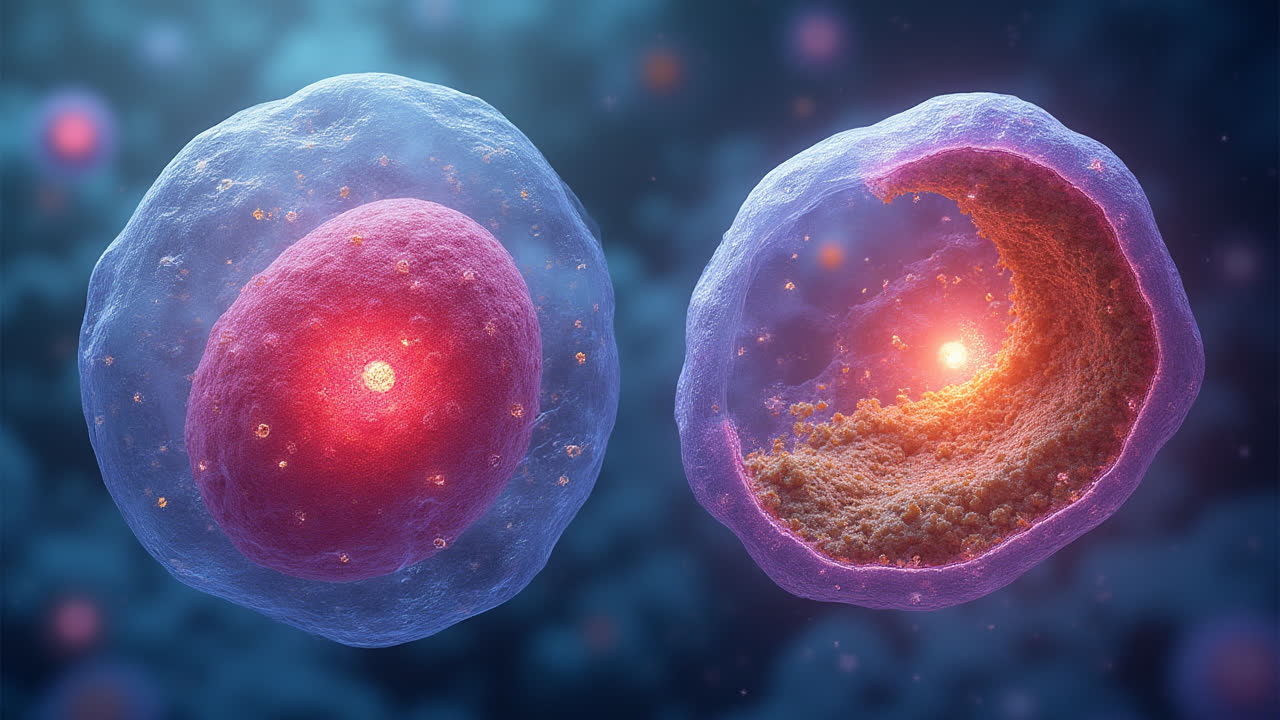
Let's start with primary lysosomes—the first stage in the lysosomal life cycle. Primary lysosomes are essentially newly formed lysosomes that haven't yet merged with material that needs to be digested. They originate from the Golgi apparatus, the cell's packaging center, which produces and packages various enzymes required for lysosomal function.
Primary lysosomes are relatively small vesicles, typically measuring about 0.1-0.5 micrometers in diameter. They're surrounded by a single phospholipid membrane that separates their acidic interior from the cytoplasm. This membrane contains specific proteins that help maintain the acidic pH necessary for enzyme function and prevent these powerful enzymes from escaping into the cytoplasm where they could cause damage.
What I find particularly interesting about primary lysosomes is that they contain hydrolytic enzymes in an inactive form. This is a crucial safety mechanism—imagine having active digestive enzymes floating around in your cell! These enzymes only become active when the lysosome fuses with another vesicle containing material to be digested, creating a secondary lysosome. This activation is triggered by the acidic environment inside the lysosome, which provides the optimal conditions for these enzymes to function.
In terms of appearance, primary lysosomes typically look like small, dense, homogeneous vesicles when viewed under an electron microscope. They contain about 50 different types of hydrolytic enzymes, including proteases, nucleases, glycosidases, lipases, phospholipases, phosphatases, and sulfatases—essentially a complete toolkit for breaking down all major classes of biological macromolecules.
The main role of primary lysosomes is to serve as reservoirs of digestive enzymes, ready to fuse with vesicles containing materials that need to be broken down. Once this fusion occurs, they transition to the next stage of their life cycle—secondary lysosomes.
Secondary Lysosomes: Formation and Activity
When primary lysosomes encounter material that needs to be digested, a remarkable transformation occurs—they fuse with vesicles containing this material to form secondary lysosomes. This fusion event marks the transition from an inactive storage vesicle to an active digestive compartment. I've always found this process fascinating because it represents such an elegant solution to the problem of controlled digestion within cells.
Secondary lysosomes form when primary lysosomes fuse with endosomes—vesicles that bring material into the cell. These endosomes can be phagosomes (containing large particles like bacteria or cell debris that have been "eaten" by the cell) or pinosomes (containing extracellular fluid and dissolved substances). The fusion process is carefully regulated by various proteins, including SNARE proteins and Rab GTPases, which ensure that lysosomes only fuse with the appropriate targets.
Once formed, secondary lysosomes are significantly larger than primary lysosomes, often appearing as irregular-shaped vesicles filled with partially digested material when viewed under a microscope. Sometimes I like to think of them as little "stomachs" where the actual digestive work of the cell happens. The previously inactive enzymes from the primary lysosome become activated in the acidic environment of the secondary lysosome, beginning the process of breaking down the internalized material.
What happens inside a secondary lysosome is quite remarkable. Complex macromolecules are broken down into their building blocks—proteins into amino acids, nucleic acids into nucleotides, complex carbohydrates into simple sugars, and lipids into fatty acids and glycerol. These building blocks can then be transported across the lysosomal membrane back into the cytoplasm where they can be reused by the cell. It's a sophisticated recycling system that operates continuously in our cells.
After digestion is complete, secondary lysosomes can take different paths. They may release their contents to the outside of the cell through a process called exocytosis, particularly if they contain indigestible material. Alternatively, they may become residual bodies containing undigested material, or they may return to a resting state similar to primary lysosomes. This cycle of lysosomal activity is essential for cellular homeostasis and adaptability.
Key Differences Between Primary and Secondary Lysosomes
Now that we've explored both primary and secondary lysosomes individually, let's directly compare these two stages of lysosomal development. Understanding these differences helps clarify how these organelles function within the cellular environment. I've compiled this comparison based on years of studying cellular biology and specifically focusing on organelle function.
| Characteristic | Primary Lysosomes | Secondary Lysosomes |
|---|---|---|
| Origin | Bud directly from Golgi apparatus | Formed by fusion of primary lysosomes with endosomes |
| Size | Small (0.1-0.5 μm diameter) | Larger, variable in size |
| Enzyme Status | Contain inactive digestive enzymes | Contain active digestive enzymes |
| Contents | Only hydrolytic enzymes | Hydrolytic enzymes plus material to be digested |
| Digestive Activity | No digestion occurs | Active digestion of materials |
| Appearance | Homogeneous, dense vesicles | Heterogeneous with partially digested material |
| Membrane Properties | Single phospholipid layer | Single membrane with specific transport proteins |
| Exocytosis Capability | Unable to eliminate contents externally | Can eliminate contents through exocytosis |
Similarities Between Primary and Secondary Lysosomes
While we've focused on the differences between primary and secondary lysosomes, it's worth noting that they share several important characteristics. Both types are fundamental components of the lysosomal system and work together to maintain cellular health. Sometimes I think we focus too much on differences without appreciating the continuity between these stages.
Both primary and secondary lysosomes are membrane-bound organelles that contain hydrolytic enzymes. They share the same basic structural organization—a single phospholipid bilayer enclosing an acidic lumen. This acidic environment is maintained by similar proton pumps in both types of lysosomes.
Additionally, both primary and secondary lysosomes contain similar sets of hydrolytic enzymes, though these enzymes are in different states of activity. The enzymes themselves—various proteases, lipases, and other hydrolases—are identical in both types of lysosomes. It's primarily their activation state that differs.
Another similarity is their role in cellular digestion. Though they represent different stages in the process, both types are part of the same digestive system of the cell. They work together in a coordinated fashion to ensure that materials are properly broken down and recycled.
Both types of lysosomes also share common regulatory mechanisms. Their formation, maintenance, and function are controlled by similar genetic and cellular pathways. Mutations affecting these pathways can lead to lysosomal storage diseases, regardless of whether they primarily affect the formation of primary lysosomes or the functioning of secondary lysosomes.
Importance in Cellular Health and Disease
The proper functioning of both primary and secondary lysosomes is absolutely crucial for cellular health. When lysosomes don't work correctly, it can lead to a range of serious conditions known as lysosomal storage diseases. I've seen firsthand how devastating these diseases can be, affecting everything from physical development to neurological function.
Lysosomal storage diseases occur when specific lysosomal enzymes are deficient or dysfunctional, leading to the accumulation of undigested materials within cells. For example, Tay-Sachs disease results from a deficiency in the enzyme hexosaminidase A, leading to the buildup of gangliosides in the brain. Gaucher disease involves a deficiency of glucocerebrosidase, causing lipid accumulation in various tissues. These conditions highlight the critical importance of functional lysosomes for normal cellular metabolism.
Beyond their role in these specific diseases, lysosomes are increasingly recognized as key players in many cellular processes. They're involved in nutrient sensing and signaling, helping cells respond to changing environmental conditions. During starvation, for instance, lysosomes play a crucial role in autophagy—the process by which cells digest their own components to recycle nutrients.
Lysosomes also play important roles in immune function. When immune cells engulf pathogens, lysosomes fuse with the resulting phagosomes to destroy the invaders. This process, where primary lysosomes transform into secondary lysosomes, is essential for our body's defense against infection.
Recent research has also implicated lysosomal dysfunction in age-related diseases such as Alzheimer's, Parkinson's, and certain types of cancer. As we age, lysosomal function tends to decline, potentially contributing to the cellular damage and dysfunction observed in these conditions. This emerging understanding of lysosomes in aging and disease has sparked interest in developing therapies targeting lysosomal function.
Frequently Asked Questions About Lysosomes
What happens if lysosomal enzymes leak into the cytoplasm?
If lysosomal enzymes leak into the cytoplasm, they can cause significant damage to cellular structures. However, this risk is minimized by several protective mechanisms. First, lysosomal enzymes are typically active only in the acidic environment of the lysosome (pH around 4.5-5.0) and are less active at the neutral pH of the cytoplasm (around 7.2). Second, the cytoplasm contains enzyme inhibitors that can neutralize lysosomal enzymes if leakage occurs. Despite these protections, severe damage to lysosomes can lead to enzyme leakage and potentially trigger cell death pathways, including a process called lysosomal membrane permeabilization, which plays a role in certain types of cell death and disease states.
Can plant cells have lysosomes?
Plant cells don't have true lysosomes as found in animal cells, but they do have structures with similar functions. Plant cells contain vacuoles, which are large organelles that can occupy up to 90% of the cell volume and perform many of the digestive and waste storage functions that lysosomes do in animal cells. Plant vacuoles contain hydrolytic enzymes similar to those found in lysosomes and can break down macromolecules. Additionally, plant cells have structures called lytic vacuoles that are functionally analogous to lysosomes. These structures help plants digest and recycle cellular components, particularly during processes like leaf senescence (aging and death). So while plants may not have organelles classified as lysosomes, they've evolved parallel structures to fulfill similar cellular needs.
How do lysosomes contribute to autophagy?
Lysosomes play a central role in autophagy, the process by which cells break down and recycle their own components. During autophagy, a double-membrane structure called an autophagosome forms around cellular components targeted for degradation, such as damaged organelles or protein aggregates. This autophagosome then fuses with a primary lysosome, creating an autolysosome (a type of secondary lysosome). The lysosomal enzymes inside this structure break down the captured cellular components into their basic building blocks, which can then be recycled by the cell. Autophagy increases during periods of cellular stress, such as nutrient deprivation, and helps cells survive by recycling non-essential components. This process is particularly important for removing damaged organelles and protein aggregates that might otherwise be toxic to the cell. Defects in autophagy have been linked to neurodegenerative diseases, cancer, and aging, highlighting the importance of lysosomal function in cellular health maintenance.
Conclusion
Understanding the difference between primary and secondary lysosomes provides crucial insights into cellular digestion and recycling processes. Primary lysosomes, those small vesicles budding from the Golgi apparatus with their inactive enzymes, represent the potential for digestive activity. Secondary lysosomes, formed when primaries fuse with endosomes, are where the actual digestive action happens—breaking down everything from ingested nutrients to worn-out cellular components.
The transformation from primary to secondary lysosomes exemplifies the dynamic nature of cellular organelles. Rather than static structures, they're constantly changing, fusing, and adapting to meet the cell's needs. This relationship between primary and secondary lysosomes demonstrates how cells compartmentalize different functions to maintain organization and efficiency.
As we continue to research these fascinating organelles, we're discovering their involvement in increasingly diverse cellular processes. From their roles in development and immunity to their implications in aging and disease, lysosomes are proving to be far more than simple "waste disposal" units. They're sophisticated regulators of cellular health with therapeutic potential we're only beginning to explore.
The next time you hear about lysosomes in a biology class or medical context, remember that these tiny organelles—in both their primary and secondary forms—are working tirelessly in nearly every cell of your body, maintaining the delicate balance necessary for life. Their proper function is not just a biological curiosity but an essential requirement for cellular health and, by extension, our overall wellbeing.